Article | 19 May 2022
The new regulation on in vitro diagnostic medical devices becomes applicable

After several years of waiting, the new EU regulation on in-vitro diagnostic medical devices will now become applicable. In this article, we provide an overview of IVDR implementation and compliance.
In 2017, two new regulations on medical devices formally entered into force, Regulation 2017/745 on medical devices (MDR) and Regulation 2017/746 on in vitro diagnostic medical devices (IVDR). The purpose of the new regulations is to promote cross-border trade and innovation and to strengthen patient safety. The application date for MDR had to be postponed due to the pandemic, but on 26 May 2021, MDR began to be applied, which we have previously reported on. On 26 May 2022, it is time for IVDR to start applying.
As a result, after 26 May 2022, IVD products may only be placed on the EU market if they meet the requirements of IVDR or are subject to the transitional provisions of IVDR, which specify how long products may continue to be placed, and made available, on the market under the requirements of the IVD Directive (IVDD). Products that are subject to the transitional provisions and may continue to be placed on the market are called legacy products or directive products.
In the table below, the Swedish Medical Products Agency has summarized the important dates for the transition to IVDR.
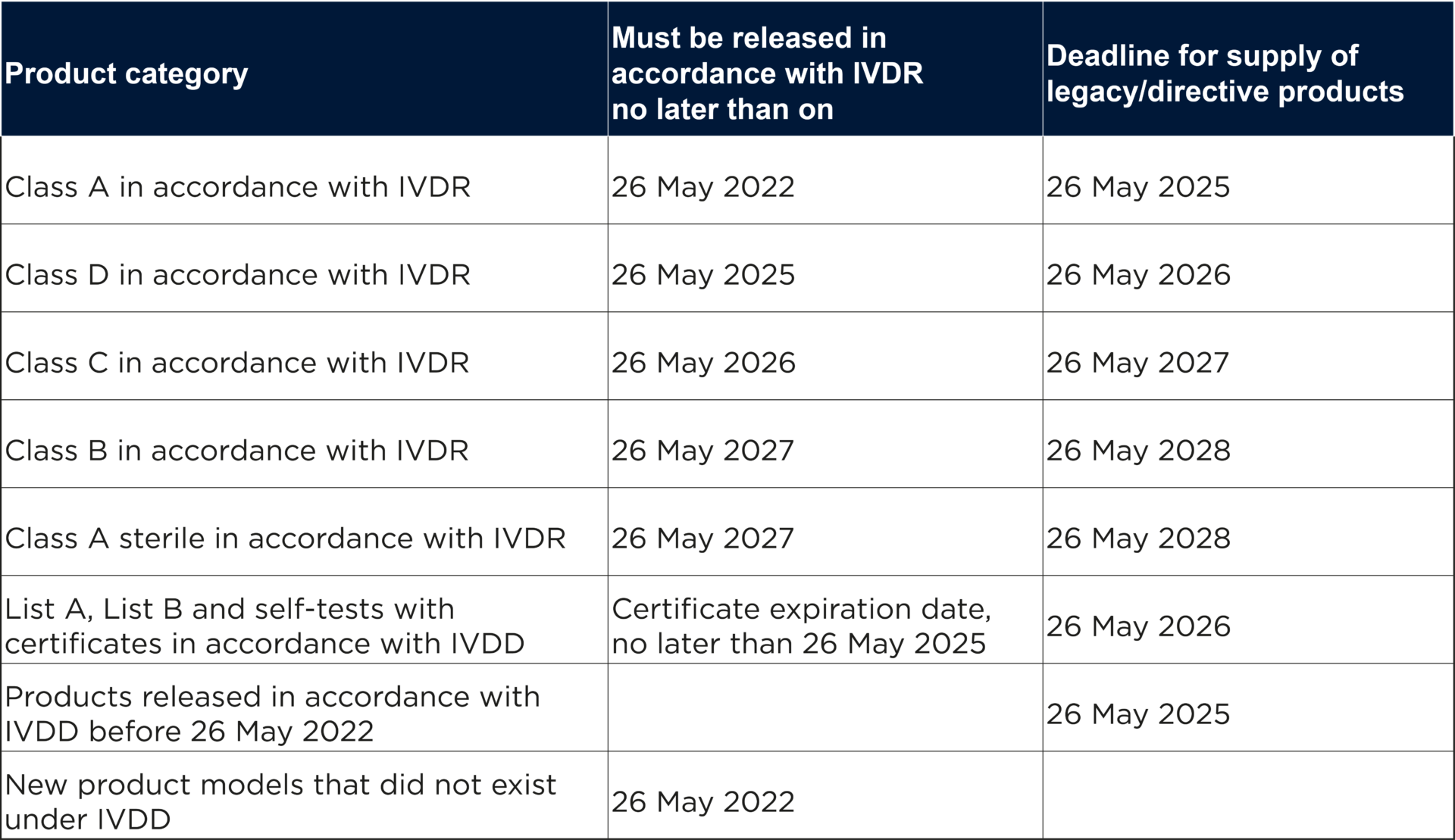
However, IVDR requires that legacy products must continue to comply with the requirements of the directive applicable to the product and that no significant changes have been made to the product’s design and intended purpose. Furthermore, the rules on monitoring of products placed on the market, market control, safety monitoring and registration of economic operators and products in IVDR must be complied with for all legacy products as early as 26 May 2022.
On 26 May 2022, the rules on unique product identification (UDI) will apply to all products placed on the market in accordance with IVDR. This means that it becomes mandatory for these products to have a unique product identification, so-called UDI, for the product.
Work on the European database Eudamed, where, for example, operators and products will be registered, is not yet complete. This means that until Eudamed is fully operational, it is the national authority (in Sweden the Medical Products Agency) to which such reports must be made.
Setterwalls will follow the work with Eudamed and other updates on the area!


